Disease information
The cell and the role of lysosomes
Cells, the building blocks of our bodies, are composed of a fluid known as cytoplasm that is encapsulated by a membrane. The cytoplasm contains the cell organs, or organelles, which have different roles in the cell’s functions and thereby keep our bodies working. Lysosomes are organelles that are responsible for breaking down many kinds of molecules (e. g. lipids, sugars, proteins) by using tools called enzymes.
The lysosomes use the enzymes to digest substances that have completed their jobs and are no longer useful, thereby cleaning the cell and preventing the accumulation of “waste” or left-over material that can impair the cell’s function. Simply said: the lysosome is the garbage man who uses enzymes to clean up the garbage… Thus, lysosomes are often referred to as the digestive track of the cell.
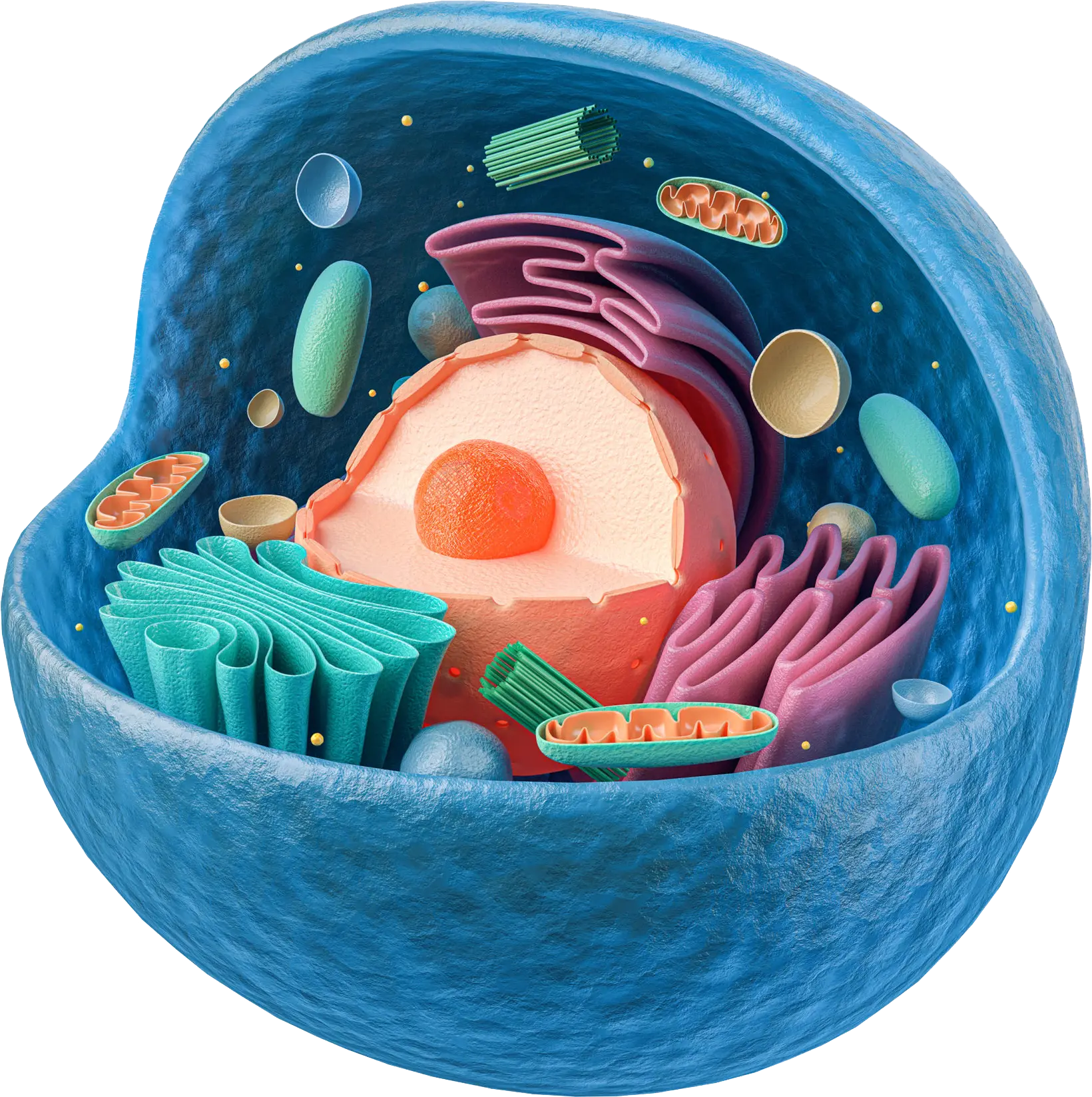
Lysosomal storage diseases
In lysosomal storage diseases, a lack or shortage of a specific enzyme due to a rare, inherited gene mutation prevents the disposal of “waste” molecules, ultimately leading to an abnormal build-up of certain “left-over” materials. This toxic accumulation can result in cell dysfunction and cell death. There are as many as 50 different lysosomal storage diseases that can affect specificareas or tissues of the body, where the accumulation takes place. These diseases are rare, inherited metabolic disorders that can cause a wide range of symptoms in various parts of the body, including the bones, heart and central nervous system (CNS).
Lysosomal storage diseases include, for example, Pompe disease, Fabry disease, Krabbe disease, Gaucher disease, Niemann Pick disease or GM1 and GM2 gangliosidoses.
GM1 Gangliosidosis
GM1 gangliosidosis is an inherited lysosomal storage disorder that is caused by a mutation in the GLB1 gene, resulting in a deficiency of an enzyme called beta-galactosidase-1. This enzyme plays an important role in breaking down large sugar molecules within the cells and its deficiency can progressively harm nerve cells in the brain and the spinal cord. The prevalence of the disease is estimated to be about 1 in 100,000 – 200,000 newborns, which ranks it as an (Ultra) Rare Disease.
Researchers have classified this condition into three major types depending on the age the symptoms start appearing:
- Type I or infantile form
- Type II or late infantile/juvenile form (disease manifestation >18 months)
- Type III or adult/chronic form (disease manifestation >5 years)
The severity of symptoms and the time at which symptoms appearcan differ greatly. However, earlier onset is often associated with severe and rapidly progressing symptoms of the disease. Currently, the treatment of GM1 gangliosidosis focuses on managing symptoms (palliative care), as there are no approved disease modifying treatments available.
Depending on the classification system, symptoms of GM1 gangliosidosis are:
- Poor muscle tone
- Enlargement of the liver and spleen (Type1)
- Skeletal abnormalities
- Enlarged gums and typical facial features (Type 1)
- Cherry red spot in the eye
- Exaggerated startle reaction
- Seizures
- Visual impairment/blindness
- Hearing loss
- Halt in motor development
- Development regression and intellectual disability
- Slowing of growth
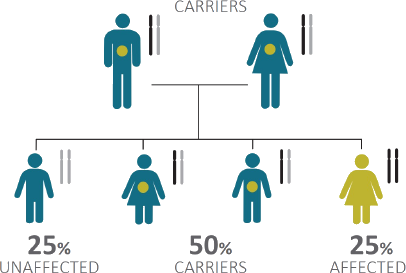
Gangliosidosis Inheritance Tree
GM1 and GM2 gangliosidoses are inherited in an autosomal recessive manner, which requires the presence of two copies of the gene mutation to express the disorder. This is the case when both parents are carriers by having a working and a non-working copy of the affected gene pair.
Graphic by courtesy of the National Tay-Sachs & Allied Diseases Association
GM2 Gangliosidosis
GM2 gangliosidoses comprise three sub-forms: Tay-Sachs disease, Sandhoff disease and AB variant. Tay-Sachs disease is named after Warren Tay, who described a patient with a red spot on the retina, as well as Bernard Sachs, who provided the first description of the changes in the cell that the disease causes. It affects 1 in 320,000 children. Sandhoff disease and the AB variant show similar clinical symptoms as Tay-Sachs disease but have different underlying enzyme mutation variants.
In GM2 gangliosidoses, patients lack sufficient levels of the enzyme Hexosaminidase that is responsible for degrading certain fats such as the GM2 molecule. Therefore, the patient has a harmful accumulation GM2 “waste” in the cell. Ultimately, this pathological storage of fats impairs cell function and can lead to cell death in the brain and other organs of the body.
At birth, children with GM2 gangliosidoses typically show no signs and develop normally during the first few months with disease onset starting typically around 3-6 months of age.
The main symptoms of GM2 gangliosidoses are:
- Cherry red spot in the eye
- Poor muscle tone
- Exaggerated startle reaction
- Seizures
- Visual impairment/blindness
- Hearing loss
- Plateau of motor development
- Development regression and intellectual disability
- Slowing of growth
Currently there is no authorized curative therapy for GM2 gangliosidosis. Patients can be treated with supportive care to ease symptoms and increase quality of life, but there remains a high unmet need for a disease-modifying treatment.
GM1 | GM2 | ||
|---|---|---|---|
Tay-Sachs disease | Sadhoff disease | ||
Gene affected | GLB1 (3p22.3) | HEXA (15q23) | HEXB (5q13.3) |
Defective enzyme | β-Galactosidase | Hexominidase A (αβ) | Hexominidase A (αβ) Hexominidase B (ββ) |
Lipid storage | GM1, lysoGM1 | GM2, lysoGM2 | GM2, lysoGM2 |
Birth prevalence | 0.26-0.62 per 100’000 Brazil 6 per 100’000 Malta 27 per 100’000 | 0.23-3.13 per 100’000 β-hexosaminidase (αβ or ββ) | 0.23-3.13 per 100’000 β-hexosaminidase (αβ or ββ) |
Niemann-Pick Type C
Patients with the lysosomal disorder Niemann-Pick disease type C (NP-C) suffer from their body’s inability to transport cholesterol and other fatty subtances inside of cells, leading to the abnormal accumulation of these lipids in varius tissies of the body, including the brain. NP-C is highly variable and can range from a fatal disorder within the first few months after birth, to a late onset, chronic progressive disorder that remains undiagnosed well into adulthood. Most cases are detected during childhood and progress to cause life-threatening complications by the second or third decade of life. NP-C is caused by mutations in the NP-C1 gene (NP-C type 1C) or the NP-C2 gene (NP-C type 2C) and is inherited in an autosomal recessive manner.
Symptoms may include:
- enlarged liver and spleen (hepatosplenomegaly)
- difficulty coordinating movement (ataxia)
- abnormal eye movements (vertical supranuclear gaze palsy)
- poor muscle tone (hypotonia)
- severe liver disease
- frequent respiratory infections
- difficulty with speech
- difficulty with swallowing and feeding
- loss of cognitive skills
- seizures
There are currently no approved therapies to reverse the effects of NP-C. Current approaches are focused on supportive therapies and targeted management for specific symptoms.